Cómo se fabrican los medicamentos genéricos: proceso explicado paso a paso
 nov, 16 2025
nov, 16 2025
Si alguna vez has comprado un medicamento genérico, probablemente te preguntaste: ¿cómo es posible que sea tan barato y funcione igual que el de marca?. La respuesta no está en trucos ni en recortes de calidad, sino en un proceso riguroso, altamente regulado y técnicamente complejo que garantiza que cada pastilla, cápsula o inyección genérica sea tan segura y efectiva como su equivalente de marca. Aunque los genéricos cuestan hasta un 85% menos, su fabricación no es más sencilla: requiere años de investigación, cientos de pruebas y instalaciones que cumplen normas tan estrictas como las de los medicamentos originales.
El punto de partida: entender el medicamento de referencia
Todo comienza con el medicamento de marca, al que se le llama Reference Listed Drug (RLD). Los fabricantes de genéricos no inventan nada nuevo: lo que hacen es desarmar el producto original para entenderlo hasta el último detalle. Esto incluye identificar el ingrediente activo (la sustancia que cura o alivia), los excipientes (los componentes inactivos como rellenos, colorantes o conservantes), y cómo están estructurados en la formulación final -por ejemplo, si la pastilla está diseñada para liberar el fármaco lentamente o en un lugar específico del intestino. Este análisis no es solo observación visual. Se usan técnicas avanzadas como cromatografía, espectroscopía y pruebas de disolución para medir exactamente cómo se comporta el medicamento en el cuerpo. Sin esta información precisa, no se puede replicar su efecto. Y aquí está la clave: el genérico no necesita repetir los ensayos clínicos que ya hizo la farmacéutica original. Eso lo permite la ley Hatch-Waxman de 1984, que creó el camino del Abbreviated New Drug Application (ANDA) en Estados Unidos. En resumen: si puedes demostrar que tu versión es bioequivalente, puedes venderla sin volver a probarla en miles de pacientes.El diseño de la fórmula: calidad por diseño
Una vez que se conoce el producto original, empieza la fase de desarrollo de la fórmula. Aquí se aplica el principio de Quality by Design (QbD), un enfoque global aceptado por la ICH (Consejo Internacional de Armonización). En lugar de probar hasta que algo funcione, se diseña desde el principio para que funcione siempre. Se identifican tres elementos críticos:- Attributos de Calidad Críticos (CQAs): características del medicamento que afectan su seguridad o eficacia -por ejemplo, la velocidad con la que se disuelve en el estómago.
- Attributos de Materiales Críticos (CMAs): propiedades de las materias primas, como el tamaño de partícula del ingrediente activo o la pureza del lactosa.
- Parámetros del Proceso Críticos (CPPs): variables durante la fabricación, como la temperatura de secado o la presión de compresión.
La fabricación: siete pasos, cero errores
La producción real sigue un proceso muy controlado, dividido en siete etapas:- Formulación: Se pesan y mezclan con precisión el ingrediente activo y los excipientes. Cada gramo debe estar dentro de tolerancias de ±0.5%.
- Mezcla y granulación: Los polvos se humedecen ligeramente y se convierten en gránulos para que fluyan bien durante la compresión. La homogeneidad es vital: si una pastilla tiene más ingrediente activo que otra, puede ser peligrosa.
- Secado: Se elimina el exceso de humedad en hornos controlados. La humedad residual no debe superar el 2% en la mayoría de los casos, o el medicamento se degradará con el tiempo.
- Compresión y encapsulación: Los gránulos se comprimen en pastillas con máquinas que aplican hasta 10 toneladas de presión, o se colocan en cápsulas. La variación de peso por pastilla debe ser menor al 5% si pesa menos de 130 mg, o al 7.5% si está entre 130 y 324 mg, según la FDA.
- Recubrimiento: Muchas pastillas reciben una capa protectora que puede disfrazar el sabor, proteger el ingrediente activo del ácido estomacal, o controlar su liberación. El recubrimiento debe ser uniforme y resistente a la humedad.
- Control de calidad: Cada lote pasa por más de 20 pruebas: identidad, potencia, pureza, disolución, uniformidad y estabilidad. Se analizan muestras en laboratorios certificados con equipos que detectan impurezas en partes por millón.
- Embalaje y etiquetado: Las pastillas se colocan en blister o frascos con etiquetas que deben coincidir exactamente con el medicamento de referencia en información de uso, advertencias y dosis -aunque pueden diferir en color o forma, por ley no pueden parecerse visualmente al producto de marca.
Las instalaciones: un entorno casi espacial
No basta con tener buenas recetas. El lugar donde se fabrica es tan importante como la fórmula. Las fábricas de genéricos deben cumplir con las Current Good Manufacturing Practices (CGMP), normas que regulan desde el aire que respiras hasta la ropa que te pones. Las áreas de producción tienen salas limpias con clasificación ISO 5 a ISO 8, lo que significa que el aire contiene menos de 3,5 millones de partículas por metro cúbico (en comparación, una habitación normal tiene más de 10 millones). La temperatura se mantiene entre 20 y 25 °C y la humedad entre 45 y 65%. El personal usa batas, mascarillas y gorros desechables, y pasa por cámaras de aire antes de entrar. Los equipos también están calibrados y validados. Una máquina de compresión no solo debe funcionar: debe demostrar que produce pastillas consistentes durante miles de ciclos. Todo se documenta: cada paso, cada ajuste, cada error. Si una pastilla pesa 10 mg más de lo permitido, se investiga en menos de 24 horas.La aprobación: el ANDA y la bioequivalencia
Antes de que un genérico llegue a las farmacias, debe pasar por la aprobación regulatoria. En Estados Unidos, eso significa presentar un ANDA a la FDA. Este documento no es un simple formulario: suele tener entre 5,000 y 10,000 páginas de datos técnicos, incluyendo:- Resultados de 200 a 300 métodos analíticos
- 10 a 15 registros de lotes de producción
- 3 a 5 estudios de bioequivalencia
Desafíos reales: lo que no te cuentan
Aunque la mayoría de los genéricos son seguros y efectivos, no todo es perfecto. Algunos desafíos persisten:- Complejidad creciente: Hoy, 35% de los ANDA pendientes son para productos complejos -como inhaladores, cremas o formulaciones de liberación prolongada- donde la bioequivalencia no se puede medir solo con sangre. Se necesitan pruebas de piel, pulmón o intestino, y eso lleva años.
- Proveedores externos: El 78% de los ingredientes activos mundiales vienen de China e India. Un cambio en la calidad de un material puede afectar miles de lotes. En 2021, Teva tuvo que retirar 14 productos por violaciones de CGMP en su planta de Puerto Rico.
- Precios bajos y márgenes ajustados: Para genéricos simples, en dos años pueden caer hasta un 80% en precio. Muchas empresas pequeñas ya no pueden competir y salen del mercado.
¿Son realmente iguales?
La respuesta corta: sí. La FDA y la Organización Mundial de la Salud coinciden: los genéricos aprobados son tan efectivos y seguros como los de marca. Un estudio de la Asociación de Medicamentos Accesibles encontró que el 89% de los farmacéuticos confían plenamente en su calidad. No todos los pacientes notan diferencias, pero algunos con enfermedades crónicas (como epilepsia o trastornos de tiroides) pueden ser más sensibles a pequeñas variaciones en la absorción. Por eso, si cambias de genérico y sientes algo distinto, habla con tu médico. Pero no lo atribuyas automáticamente a la calidad del medicamento: a veces, es solo el cuerpo adaptándose.El futuro de los genéricos
El mercado global de genéricos crece un 6-8% anualmente. Para 2027, más de $75,000 millones en medicamentos de marca perderán su patente, incluyendo fármacos clave como Eliquis y Stelara. Esto abrirá oportunidades, pero también presiones. Las tendencias más importantes son:- Genéricos autorizados: las mismas empresas de marca que lanzan su propia versión genérica, aprovechando su conocimiento.
- Manufactura digital: sistemas de inteligencia artificial que detectan defectos en pastillas en tiempo real, reduciendo errores de inspección visual en hasta un 40%.
- Biológicos genéricos: versiones de medicamentos hechos con células vivas, como los tratamientos para el cáncer o la artritis. Son más complejos, pero ya están llegando.
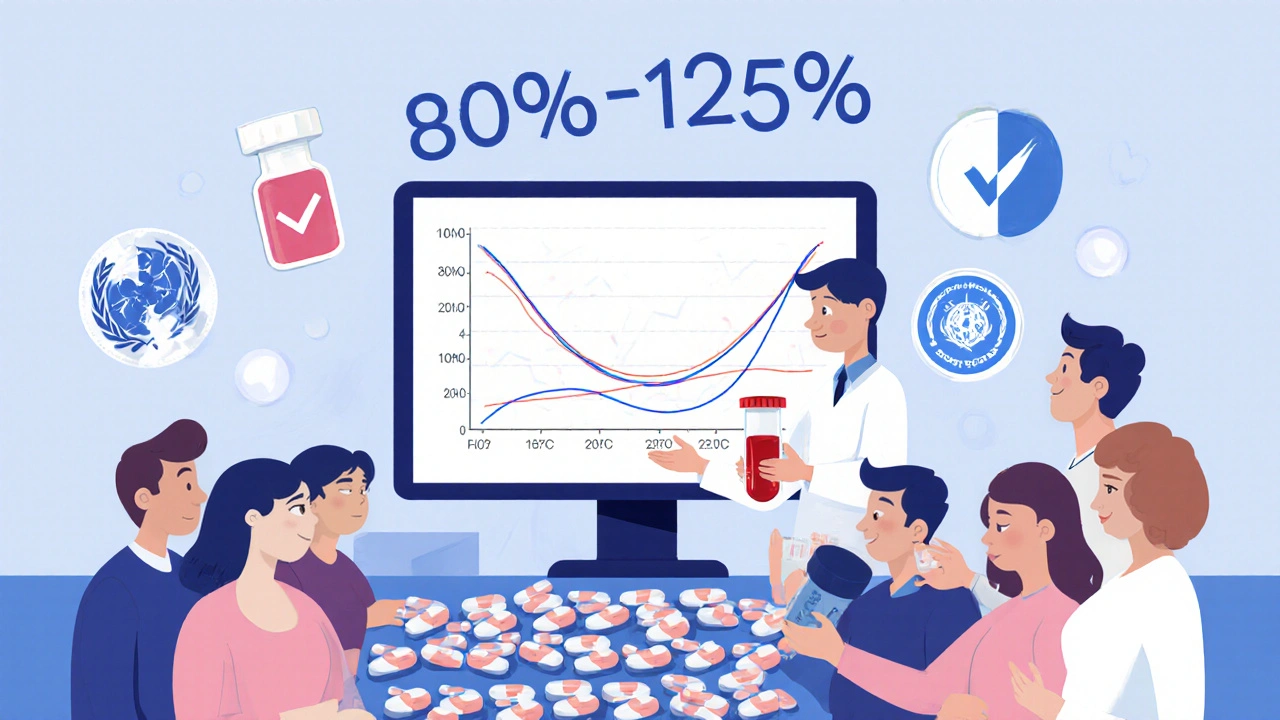
¿Los medicamentos genéricos son tan efectivos como los de marca?
Sí. La FDA exige que los genéricos tengan el mismo ingrediente activo, dosis, forma, vía de administración y eficacia que el medicamento de marca. Para aprobarse, deben demostrar bioequivalencia: su absorción en el cuerpo debe estar entre el 80% y el 125% del original. Estudios clínicos y revisiones de la OMS confirman que no hay diferencias significativas en resultados de salud.
¿Por qué los genéricos son más baratos?
Porque no tienen que repetir los costosos ensayos clínicos que ya hizo la farmacéutica original. El proceso de aprobación (ANDA) se basa en los datos de seguridad y eficacia del medicamento de marca. Además, al haber múltiples fabricantes compitiendo por el mismo producto, los precios bajan naturalmente. El ahorro no viene de recortar calidad, sino de evitar duplicar gastos innecesarios.
¿Pueden los genéricos tener diferentes efectos secundarios?
Los efectos secundarios vienen del ingrediente activo, que es idéntico. Pero los excipientes (como colorantes o conservantes) pueden variar. En raras ocasiones, alguien puede tener una reacción leve a un compuesto inactivo diferente -por ejemplo, a un colorante o a un agente de relleno como el gluten. Si tienes alergias o intolerancias, revisa la lista de ingredientes en el prospecto. Si notas algo nuevo después de cambiar de genérico, consulta a tu médico.
¿Qué significa que un genérico sea "complejo"?
Un genérico complejo es aquel que no se puede comparar fácilmente con una simple prueba de sangre. Incluye inhaladores, cremas tópicas, inyecciones de liberación prolongada o soluciones oculares. En estos casos, la forma física y cómo se libera el fármaco en el cuerpo son tan importantes como la cantidad. Por eso, desarrollarlos lleva más tiempo, más dinero y más pruebas técnicas. Solo el 12% de los genéricos eran complejos en 2015; hoy son el 35%.
¿Son seguros los genéricos fabricados en China e India?
Sí, siempre que cumplan con las normas de la FDA o la EMA. La FDA inspecciona todas las fábricas que exportan medicamentos a Estados Unidos, sin importar su ubicación. En 2023, más del 78% de los ingredientes activos venían de China e India, pero las instalaciones que no cumplen son cerradas o suspendidas. Los problemas de calidad no están ligados al país, sino a la empresa y su gestión. Un buen fabricante en India puede ser más confiable que uno mal gestionado en Estados Unidos.
Bella Nira Aparicio
noviembre 17, 2025 AT 05:30Me encantó este artículo. Por fin entiendo por qué mi médico insiste en los genéricos. No es por ahorrar dinero, es por confiar en la ciencia.
Gracias por explicarlo tan claro.
Carlos Flores
noviembre 18, 2025 AT 05:14El nivel de rigor técnico descrito aquí es abrumadoramente impresionante. Se requiere una disciplina casi monástica para producir un genérico conforme a las normas internacionales. La industria farmacéutica global debería rendir homenaje a estos ingenieros anónimos que garantizan la salud pública con precisión milimétrica.
Cristian Falcon
noviembre 20, 2025 AT 02:06Lo que más me sorprende es que todo esto sea invisible para el consumidor. Nadie ve las salas limpias, los análisis de ppm, los 200 métodos analíticos. Solo vemos una caja más barata. Y eso es lo bonito: la calidad que no se ve es la que más cuenta.
La ciencia callada.
alexandria romero
noviembre 22, 2025 AT 00:09¿Y si el excipiente es diferente? ¿Eso realmente no afecta?
Ramon Villain
noviembre 23, 2025 AT 22:20Yo tengo un familiar con epilepsia y cambió de genérico una vez. Le dio mareos por una semana. No fue el medicamento, fue su cuerpo ajustándose. Pero lo dijo con miedo, como si fuera culpa suya. Por eso, lo que importa no es solo la ciencia, es también educar a la gente para que no se asuste de lo que no conoce.
La confianza se construye con información, no con marketing.
raul perez
noviembre 24, 2025 AT 09:38¡¿Qué?! ¿Que el ingrediente activo es el mismo pero los excipientes cambian? ¡Eso es un fraude disfrazado de ciencia! ¿Y si el colorante es carcinogénico? ¿Y si el almidón viene de un proveedor chino que usa pesticidas? ¡La FDA no puede estar revisando todo! ¡Esto es un escándalo!
tania parra
noviembre 25, 2025 AT 20:01Me alegra que alguien haya explicado esto con tanta claridad. A veces siento que los medicamentos genéricos son vistos como de segunda clase, y eso es injusto.
La verdad es que detrás de cada pastilla hay una batalla de calidad, no de precio.
Gracias por recordarnos que la economía puede ser ética.
Luisa Avila
noviembre 27, 2025 AT 08:04¿Alguien más piensa que esto es una farsa para que las farmacéuticas grandes sigan ganando? ¿No creen que la FDA está en caja de cristal con las grandes compañías? ¿Y si los estudios de bioequivalencia se manipulan? ¿Y si los laboratorios en India están falsificando los datos? Yo no confío. Yo pido el original. Si no puedo pagar, no lo tomo. Mejor morir con dignidad que vivir con un producto sospechoso.
Laura Gutiérrez
noviembre 27, 2025 AT 12:10¡Muy buen análisis! Pero me gustaría añadir algo importante: si alguien cambia de genérico y siente algo distinto, no es culpa del medicamento. Es normal que el cuerpo se adapte a pequeñas variaciones en la formulación, incluso si son técnicamente insignificantes.
Lo que sí es crucial: no auto-diagnostiques. Habla con tu farmacéutico, pide el prospecto, compara los excipientes. Y si hay dudas, pide un informe de bioequivalencia. ¡Tienes derecho a saber! La salud no es un lujo, es un derecho. Y tú mereces información clara, no rumores.
Agustin Lopez
noviembre 27, 2025 AT 17:29En España, cuando empecé a tomar genéricos, pensé lo mismo que muchos: que era una trampa. Pero luego conocí a un farmacéutico en Sevilla que me explicó esto mismo. Me mostró los certificados de calidad, las inspecciones de la EMA... y cambié de opinión.
La cultura del medicamento de marca es una herencia del marketing, no de la ciencia.
La ciencia no tiene marca. Solo tiene resultados.
Katherine Hinojosa
noviembre 28, 2025 AT 17:26¡Este es el tipo de contenido que debería ser obligatorio en las escuelas! No solo para entender medicamentos, sino para aprender a confiar en la ciencia y no en los miedos.
¡Vamos a difundir esto! ¡Cada persona que entienda esto es una persona menos que cae en el miedo y la desinformación!
Gracias por hacer esto. ¡Sigue así!
rosa maria alonso ferragud
noviembre 29, 2025 AT 00:13¿Y qué pasa con los que no pueden pagar ni el genérico? ¿No es esto una burla? ¿Por qué no bajan los precios más? ¿Por qué no se hace algo real para que todos tengan acceso? ¿Por qué siempre hay alguien que se beneficia mientras otros sufren?
Todo esto está muy bonito, pero no cambia la realidad: si no tienes dinero, no tienes medicina. Y eso no es ciencia. Eso es injusticia.
Wendy León
noviembre 30, 2025 AT 01:11¡Ah, claro! Todo perfecto, ¿no? ¿Y las inspecciones que no se hacen? ¿Y los lotes que se venden sin revisar? ¿Y los 300 medicamentos que se retiraron en 2023 por contaminación? ¿No les parece que esto es como decir que el avión es seguro porque los pilotos tienen licencia, aunque la torre de control esté dormida?
La realidad es que nadie revisa todo. Y tú, que confías, eres el que paga el precio.
Jose Antonio Pascual
noviembre 30, 2025 AT 04:55La verdad es que este artículo es una propaganda de la industria farmacéutica disfrazada de educación. ¿Qué saben ustedes de las prácticas reales en las fábricas? ¿Han visto las denuncias de whistleblowers? ¿O solo leen los informes de la FDA, que son los mismos que aprueban las patentes que luego explotan?
Todo esto suena bien en un blog, pero en el mundo real, los genéricos son un negocio de baja calidad, alta ganancia, y mucho silencio.
Y tú, que lo crees, eres el tonto que paga por la ilusión.